Pig Genome White Paper
Submitted on behalf of the International Swine Genome Sequencing Consortium (SGSC) by
Alan L. Archibald*
Mario Caccamo*
Joe Cassady
Carol Churcher*
Daniel Ciobanu
Cathy Ernst
Merete Fredholm*
Claire Rogel-Gaillard
Martien A.M. Groenen*
Joan Lunney
Denis Milan*
James Reecy
Gary Rohrer
Max F. Rothschild*
Lawrence B. Schook*
Christopher K. Tuggle
For further information please contact either
Lawrence B. Schook (SGSC Chair) at schook@illinois.edu or
Max Rothschild (NRSP-8 Pig Genome Coordinator) at mfrothsc@iastate.edu
SGSC Steering Committee*
This document was developed in consultation with the SGSC Steering Committee that is representative of global researchers and members of the NRSP8 pig genome committee with specific input from topic leaders developing the pig genome sequence manuscript [see Archibald et al. 2010]. The document was also vetted through consultation with the broader SGSC community.
1. Current Status of genome. The sequence data from the current genome assembly (Sscrofa Build 10) has been established and is comprised of hierarchical shotgun sequence data providing 4-6x genome coverage from BAC clones (CHORI-242) representing a minimal tile path across the genome plus >30x genome coverage of whole genome shotgun sequence (WGS) data generated using Sanger (capillary) and next-gen (Illumina) technologies from the same animal (Duroc 2-14). The minimal tile path was identified from a high quality physical (BAC contig) map (Humphray et al., 2007) and provides coverage of 98.3% of this physical map.
As of July 5, 2010 the total length of the BAC-derived sequence contigs, prior to the removal of sequence redundancy between overlapping BAC clones, was 3.01 Gbp of which 156.3 Mbp was at finished quality. The BAC-derived sequence data in the current assembly were generated from 14,587 BAC clones of which 12,454 have been subjected to one round of automated pre-finishing and 951 were sequenced to fully finished standard. Summary statistics for the current assembly (Sscrofa10) are presented in Table 1 (note: details of the size and number of WGS contigs are not included; these contigs which cover the 10% of the genome missing from the BAC-derived sequences are significantly smaller).
|
Genome Length |
|
|
Total Length (chr 1-18,X,Y) |
2,540,767,118 |
|
Chr U Length |
1,419,636 |
|
Chr U WGS Length |
240,478,184 |
|
Total |
2,782,664,938 |
|
Contigs |
|
|
No. of Contigs (chr 1-18,X,Y) |
59,744 |
|
No. of contigs in U |
34 |
|
Total no of contigs |
64,362 |
|
N50 / 90 total |
97,955 / 17,151 |
|
N50 / 90 U |
68,619 / 19,616 |
|
Scaffolds |
|
|
Total length of scaffolds |
2,588,106,638 |
|
Total number of scaffolds |
8,486 |
|
Average length of scaffolds |
304,986 |
|
Largest scaffold length |
6,085,579 |
|
N50 / 90 |
806,269 / 177,619 |
The Biotechnology and Biological Sciences Research Council (BBSRC), UK is funding work by the Wellcome Trust Sanger Institute in collaboration with the University of Cambridge to sequence and annotate the pig X and Y chromosomes. This specific project aims to deliver finished sequence of the porcine X chromosome and substantial segments of the Y representing transcribed regions. Further aims of this project include a) identifying the X-Y homologous genes shared with other mammals and those specific to the pig; b) identifying amplified sequence families, their organisation on X and Y and their co-evolution in suids.
2. Need for Refinement. In addition to being important for the world’s continued food supply, the pig is an important model organism for biomedical research. A reliable assembly of the porcine genome sequence is a prerequisite both for supporting sustainable genetic improvement, and for understanding the genetic basis of phenotypic variation that can effortlessly be translated into uses for the human population. The current assembly greatly facilitates genetic studies shortening the distance between phenotype and genotype. Nevertheless, within the current assembly (build10) a considerable amount of sequence (5%) still appears to be erroneously duplicated or missing. The current assembly is based on individually sequenced BACs derived from libraries from a single Duroc sow. Both the BACs selection and placement were based on an excellent physical map [Humphray et al., 2007] where BACs covering a tile path for every fingerprint contig (FPC) were selected for sequencing. Only a limited number of species have had their genome sequenced based on a physical map. This approach provides a reliable representation of the genome architecture and chromosome placement that enables accurate large scale analyses such as genome wide association studies (GWAS), and other population-based studies. Many of the sequenced BACs, however, still consist of multiple contigs and gaps that need to be closed to, for example, remove redundant duplications. These local misassemblies have a direct impact on the quality of the annotation, and the ability to represent features such as alternative splice forms. Furthermore, the pig is an important reference genome for a large number of closely related Suids whose genomes have been sequenced at low coverage. A good comparison of the genomic changes during speciation in this group of species requires a high quality reference genome of the domestic pig.
Comparison to other species, especially at the level of gene family evolution, also requires a highly accurate sequence. Manual annotation of over 500 immune related genes has found many examples of alternative splicing, as well as clear instances of gene duplication and divergence, which is supported by mRNA evidence. These novel active genes encode proteins which appear to be specific to the pig. However, this extensive manual annotation using cDNA and genome sequence evidence has also documented many errors of assembly in the build that was available for such detailed annotation (Sscrofa9). As shown for the human finished genome in 2004, a highly accurate genome can correct such assembly errors and resolve whether new gene family members are present or a result of errors inherent in trying to assemble duplicated regions with draft sequences.
A genome sequence is an essential tool for 21st Century biological, especially genetic, research of an organism. Genome sequence assemblies, including the ‘finished’ human and mouse sequences are subject to continual revision as new data are acquired and errors corrected. New sequencing technologies are enabling the development of new high throughput assays in which the read out is sequence data (e.g. RNA-seq, ChIP-seq, methylation site mapping) as well as the sequencing of multiple individuals for a species of interest. Analysis and interpretation of these data are critically dependent upon up-to-date and well annotated reference genome sequences for the species of interest. The sequence data from these assays in turn enables ever richer annotation of the reference genome sequences.
3. Approach. The goals and approaches proposed by the Swine Genome Sequence Consortium for refinement of the pig genome sequence are modeled on those of the Genome Reference Consortium (GRC) ( http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/ ), i.e., to ensure that the pig reference assembly is biologically relevant by closing gaps, fixing errors and representing complex variation and gene structure.
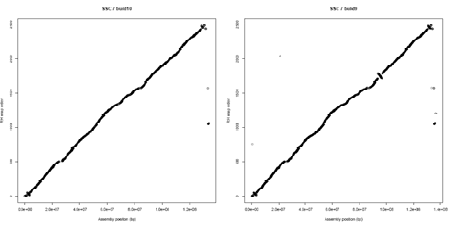
A. There are opportunities to improve the quality of the current assembly (Sscrofa10) through bioinformatics analyses of the current sequence data and other genomic information (e.g. radiation hybrid and linkage maps). The order of 34K SNPs in a high resolution radiation hybrid map has been compared with the order deduced from the current and previous assembly (Sscrofa10 and Sscrofa9) – see figure 1. Whilst the colinearity of the RH and sequence maps has improved, inconsistencies and anomalies remain. The inconsistencies will be examined and experiments designed to resolve them. For example, multicolor fluorescent in situ hybridization to interphase chromosomes using BAC clones as probes would allow the order of BAC clones in the FPC physical map and as predicted by the assembly and the RH map to be validated.
The PGP viewer (http://pgpviewer.ensembl.org/index.html) developed at the Wellcome Trust Sanger Institute will be used to visualize sequence data such as paired-end reads from Illumina WGS data, fosmid clones and BAC clones in the context of the assembly to check for consistency between the assembly and the expected orientation of paired-end sequences. The PGP viewer is based on a customized version of the Ensembl core infrastructure that also provides an interface (API) for a programmatic access of the data. The Illumina WGS data, which has been generated from the same DNA source as the CHORI-242 BAC library, provides substantially greater sequence depth (>30x) than the BAC-clone derived sequence (4-6x). Thus, there are opportunities to improve the confidence with which each base in the reference sequence has been called by comparison of the WGS and BAC-derived sequences. As the Illumina WGS data are not constrained by the cloning bias associated with the BAC and fosmid clones, there will be opportunities to characterize the “unclonable” parts of the genome, including potentially the pericentromeric sequences. It will be necessary to make appropriate allowances for the fact that the BAC-derived sequences in effect represent a haploid genome, albeit not consistently the same haploid genomes in all the clones, while the WGS data represent the diploid genome.
Figure 1.
Alignment of the RH maps and sequence assemblies Sscrofa10 and Sscrofa9 with respect to chromosome 7. While the major inconsistencies present in build 9 have been resolved other anomalies remain unresolved towards the ends of the chromosome.
Community engagement in manual annotation provides a further bioinformatics-based approach to refine and improve the genome sequence. Manual inspection of the gene models predicted by the automated annotation system, especially with reference to comparative genomic information will reveal omissions and inconsistencies not only in the gene models but also the underlying sequence. The “otterlace” system developed at The Wellcome Trust Sanger Institute is being used by the pig genome community for manual annotation. Those engaged in manual annotation will be encouraged to use the GRC-style ticket system to request corrections assembly and to commission targeted finished sequencing of genes or regions of interest. More members of the community will likely participate in such time-consuming manual annotation once a highly accurate genome is available, as the sequence will resolve important biological questions of interest to the community that are unresolved with the current data. This in turn will lead to a more highly annotated and useful genome sequence. There is considerable need for continued annotation given that we just recently finished the sequencing.
The functional analysis of the genome is the next frontier and most aspects of functional genomics involves mapping short read sequences to the genome, such as RNA sequencing to directly measure expression and map RNA structure, as well as methods such as chromatin immunoprecipitation to investigate location of regulatory signals. However, these methods require that the underlying genome sequence must be as accurate as possible to map these reads correctly. For example, placing the regulatory elements in the correct strand is a key piece of information required by the downstream analysis.
B. The hierarchical shotgun (clone-by-clone) sequencing strategy facilitates an iterative and targeted approach to improving the contiguity and coverage of the reference genome sequence. A single round of automated prefinishing (i.e. primer walking from contig ends) has been performed on most of the sequenced BAC clones. Further gap closure would be conducted by sequencing PCR fragments amplified from specific BAC clone DNA preparations. After each round of primer walking / PCR fragment sequencing opportunities for further gap closure using the Illumina WGS sequence contigs would be explored. The choices of BAC clones or regions for targeted gap closure and finishing would be determined by the community using a ticket system modeled on that employed by the Genome Reference Consortium.
The current assembly covers 98.5% of the physical map established by Humphray et al. (2007). There are some known gaps for which neither BACs nor fosmids have been sequenced. There are also a small number of BAC contigs that are not integrated into the map. Thus, in addition to sequence improvement by gap closure of sequenced BACs, additional BAC and fosmid clones will be selected for sequencing to close gaps between clones. Pools of such clones will be sequenced using next-generation sequencing technology – probably 120+ bp paired end reads using the Illumina platform. Again the Illumina WGS data will be used to assist the integration of these additional sequence data into the assembly.
3. Although a high quality reference genome sequence provides a critical framework for analysis of sequence-based assays, no single genome represents the complete repertoire of genomic information for a species. Characterizing structural variants (SVs), including insertion/deletions (indels) and copy number variants (CNVs) is particularly important in order to provide more complete sequence coverage of pig genomes. Sequencing and de novo assembly of additional individuals from other breeds would facilitate the identification of specific rearrangements and large deletions (SVs) in the genomes of pigs. Currently a large number of individuals are being sequenced, but the sequencing efforts are focusedl primarily on SNP discovery (i.e., only short insert libraries are sequenced at 8x). Extending these ongoing efforts to include mate pair libraries with larger inserts would enable the identification of SVs. De novo sequencing of additional pigs will enable the construction of contigs for regions missing in TJ Tabasco (2-14). The subjects of these re-sequencing efforts would include individuals from lines of minipigs used in biomedical research as well as commercially important breeds and material informative from an evolutionary perspective (wild boar and related suids). The overall aim is to generate a comprehensive and deep catalogue of genetic variation across the domesticated pigs and other suid species similar to the 1000 genomes effort.
4. Expected Value and Impact. The swine genome community has just recently received the sequence information and is beginning to digest and understand it. In the academic world two groups of researchers will be aided by refinements. The first group, swine geneticists, will use refinements to locate QTL for traits of economic importance and working closely with the swine industry these will translate into improved pork production with higher quality pork, better pig health and safer pork meat production. The second academic group will be primarily biologists who will use the refinements to better understand the evolutionary role of gene changes in the pig and related species. In addition, those academics with interest in human medicine will use the refinements in the pig to improve the role of the pig as a biomedical model for humans.
5. Consultation with the researchers in the pig genome community. In November 2009 the pig genome community met to announce the near completion of a draft genome sequence for the pig. This meeting and several others over the summer along with other discussions that have been held over the past year have been directed to not only the use of the sequence but also the future needs and directions which are outlined here. The plan presented here is well vetted and represents views from research not only in the US but around the world and includes the SGSC steering committee.
6. Long term disposition of the sequence and its annotation. All the sequence data on which the current assembly (Sscrofa10) have been placed in the relevant public domain databases, including NCBI Genbank, the European Nucleotide Archive (ENA), Trace repositories and Sequence Read Archives. Similarly, the assemblies, including Sscrofa10 have been deposited in Genbank and ENA. New sequence data and assemblies will be place in the appropriate public domain databases in a timely manner in accordance with the Toronto Statement on pre-publication data sharing. “The Swine Genome Sequencing Consortium is registered as ‘owner’ of the sequence on behalf of the community”. The SGSC will be responsible for authorizing changes to the reference sequence and depositing revised assemblies in the public domain. The genome sequence will be annotated using the Ensembl and NCBI automated pipelines. Funds to maintain the currency of the Ensembl annotation are being sought from the BBSRC, UK – decision expected in Spring 2011. In addition, all members of the swine research community can manually annotated swine genes using the Welllcome Trust Sanger Institute’s “Otterlace” annotation software and mount additionally annotation using the Distributed Annotation System as DAS tracks in Ensembl.
Unfortunately, there is no long term funded official Swine Genome Database. The swine community strongly supports the concept of establishing a long term “Official” Swine Genome Database. However, due to the similarity between livestock species, the swine community also recognizes the need to meet the database needs of all livestock species. Thus, the swine community is extremely supportive of the development of a Livestock Genome Database Center of Excellence. The focus of this center of Excellence should be to meet the genome database needs of the livestock community. For example, The Center would be responsible for updating and improving genome assembles and annotations, cataloging genome variants, gene nomenclature, etc. Such a Center of Excellence needs to be established as soon as possible. Open access to all such databases will be required.
Literature Cited
Archibald, A.L., Bolund, L., Churcher, C., Merete Fredholm5, Martien AM Groenen6, Barbara Harlizius7, Kyung-Tai Lee, Denis Milan, Jane Rogers10, Max F Rothschild, Hirohide Uenishi, Jun Wang, Lawrence B Schook, the Swine Genome Sequencing Consortium. 2010. Pig genome sequence - analysis and publication strategy. BMC Genomics 11:438.
Humphray SJ, Scott CE, Clark R, Marron B, Bender C, Camm N, Davis J, Jenks A, Noon A, Patel M, Sehra H, Yang F, Rogatcheva MB, Milan D, Chardon P, Rohrer G, Nonneman D, de Jong P, Meyers SN, Archibald A, Beever JE, Schook LB, Rogers J: A high utility integrated map of the pig genome. Genome Biol 2007, 8(7):R139
Toronto International Data Release Workshop Authors: Prepublication data sharing. Nature 2009, 461:168-70.
[ PDF version ]